Ribonuclease A is a 124 amino acid protein that is folded into a structure of 3 a -helices and 5 b -sheets. It is kidney-shaped and has the active site located in a deep cleft. RNase A was involved in a study that showed "it is largely a protein’s internal residues that direct its folding to the native conformation" (1). It’s folding pattern was also one of the first to be studied using 1H-NMR which also looked at pH-induced structural changes and the binding of active site inhibitors. These studies produced much of the information that we now have about the structure and function of ribonucleases.
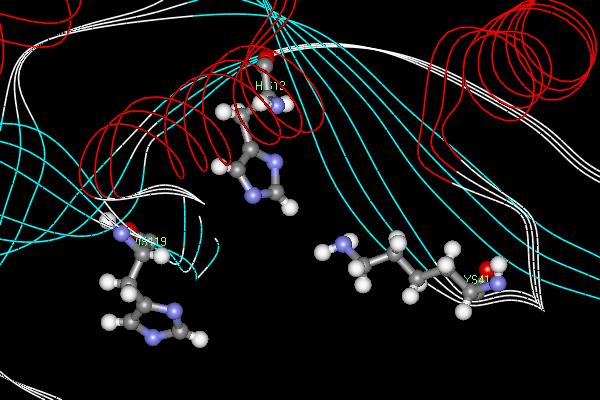
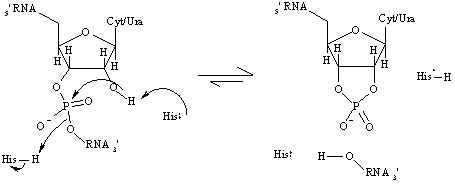
Bovine pancreatic RNase A is a well has an enzymatic mechanism dependent on general acid-base catalysis. The two most important active site residues are His 12 and His 119 which both act as proton "sinks" in the mechanism (Figure 2). The RNase reaction is a two step process whereby the P-O5’ bond in RNA is cleaved and consequently cleaving the RNA strand (1). In the first step, His 12, acting as a general base, abstracts a proton from an RNA 2’-OH group creating a stronger nucleophile which will attack the nearby phosphorous atom. Simultaneously, His 119 , acting as a general acid, protonates the leaving group which promotes bond scission. In the second step, this 2’,3’-cyclic intermediate is hydrolyzed and water replaces the leaving group and the two histidines residues reverse roles. His 12 and His 119 act as an acid and a base, respectively, leaving the enzyme in its original state.
Figure 1. Active site of the endonucleaseRibonuclease A. The two histidine residues serve as proton "sinks" while the lysine's positive charge stabilizes the negative ion of the transition state. Ribonuclease A cleaves RNA molecules after pyrimidine nucleotides.
There is also another residue whose role is not as important in the actual mechanism but functions to stabilize the transition state. Lys 41 donates a hydrogen bond to the rate-limiting transition state during catalysis and also stabilizes the excess negative charge on the oxygens during RNA cleavage (2). Researchers determined this by replacing lys 41 with other residues of similar pKa and measuring kcat, kM, and kcat/kM. The results of the experiment are summarized in Table 1. The results indicate that the binding affinity of the enzyme for the substrate does not change if residue 41 is not lysine. However, the turnover rate, kcat, is affected and the two non-wild type enzymes are not nearly as efficient as lysine, although arginine is closer than cysteine. This is probably because arginine is positively charged and has some of the same effects as lysine. The results of this experiment also show that one hydrogen bond is sufficient to effect efficient catalysis.
Residue 41
kcat (s-1)
kM (mM)
kcat/km (M-1s-1)
Table 1. kcat, kM, and kcat/kM values for different residues at position 41 in RNase A.
Figure 2. Mechanism for the cleavage of RNA as catalyzedby RNase A. The histidine residues act as proton "sinks", donating and accepting H+ atoms as necessary duringthe reaction. The histidine that donates a proton in the first step is His 119 and the histidine that accepts a proton is His 12.
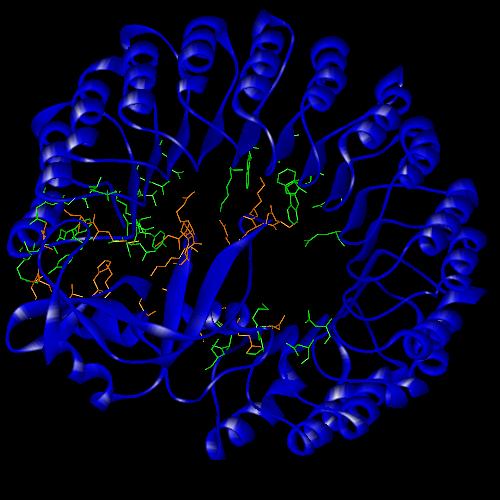
Inhibition is an important part of any enzymatic system. There have been many inhibitors of ribonucleases characterized including one category that is based on interactions between leucine-rich repeats (LRRs) and protein ligands. Porcine ribonuclease inhibitor (RI) has a primary structure composed of 15 leucine-rich repeats, alternately 28 and 29 residues long (3). The structure is non-globular and has a horseshoe-like shape in which the individual LRRs represent structural b -a hairpin units (Figure 3). The RI is much larger than RNase A with 456 residues in its structure.
When the inhibitor binds to RNase A, the active site of the enzyme is centered on the C-terminus of the inhibitor. One lobe of the RNase molecule reaches into the concave cavity of the RI molecule. This conformation prevents substrate from accessing the active site and it is therefore considered competitive inhibition. The structure of the bound RNase A is not significantly different from the structure of free RNase A. However, the inhibitor enlarges the cavity in order to accommodate the RNase molecule. There is no obvious hinge involved, small shifts accumulate along the chain and change the overall curvature and twist of the horseshoe. There are many hydrogen bonding and van der Waals interactions between the molecules, most of which are concentrated at the C-terminus of RI (Figure 3).
Figure 3. Ribonuclease A bound with Porcine Ribonuclease Inhibitor. The residues of eachmolecule that contribute to hydrogen bonding and van der Waals interactions between the two molecules are highlighted. Most interaction occurs at the C-terminus of the RI molecule. The inhibitor functions by blocking the active site of the RNase A molecule.